Density Functional Theory (DFT) calculations are one of the most accurate and expensive simulations to investigate nanomaterials. Electronic properties, such as band structure of materials are widely studied by DFT. The band structure and its change due to applied strain of an MoS2 monolayer are conducted as an example.
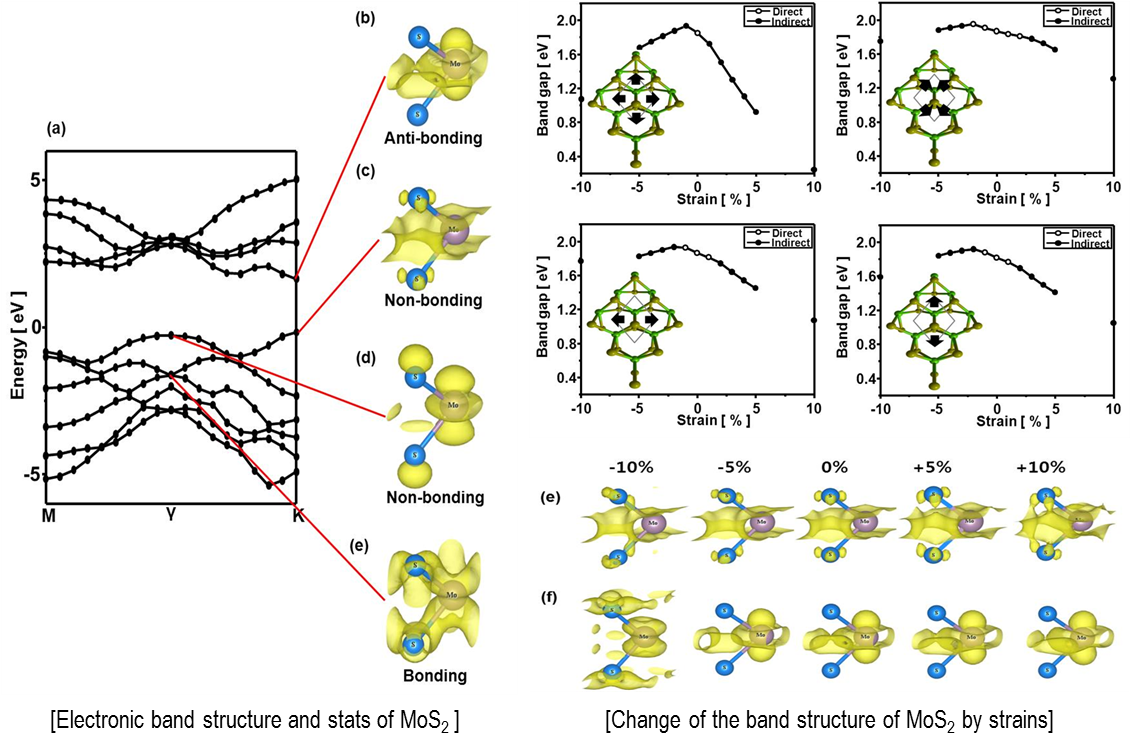
An MoS2 monolayer show direct band gap unlike its counterpart bulk. However its direct band gap easily turns to indirect band gap when small stain applied. Through DFT calculations, we can analyze the electronic structure and wave functions of each states, and thus understand basic mechanism underlying the phenomenon.